Een nieuw geneesmiddel, OTQ923, dat is ontwikkeld met behulp van de CRISPR-Cas9 gentechnologie laat goede resultaten zien in de behandeling van sikkelcelziekte. Dat is de conclusie uit een studie door een team wetenschappers aan het St Jude’s Childrens Hospital in Memphis (Verenigde Staten) onder leiding van dr. Akshay Sharma. De studie is gepubliceerd in het New England Journal of Medicine.
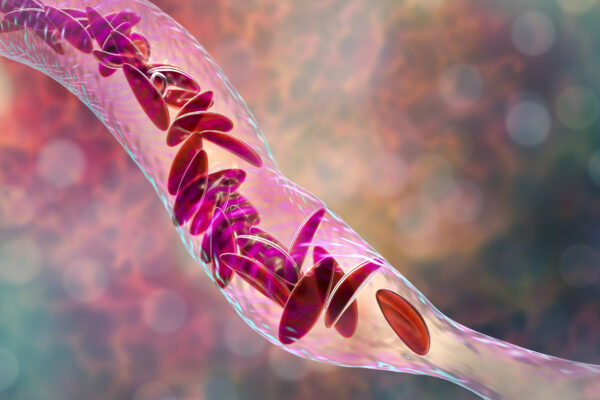
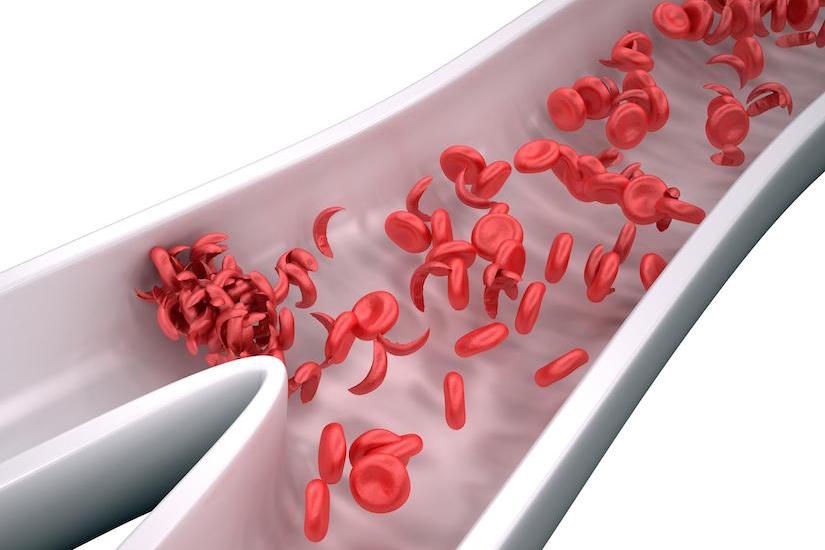
Sikkelcelziekte wordt veroorzaakt door een defect in de β-globine subeenheid van volwassen hemoglobine waardoor hemoglobine S (HbS) ontstaat. Bij verminderde zuurstof polymeriseert sikkelcel hemoglobine waardoor misvormde rode bloedcellen ontstaan die celafbraak veroorzaken en bloedvaten kunnen afsluiten met als gevolg hevige pijnaanvallen, progressieve orgaanschade en vroegtijdige dood. Het is bekend dat verhoogde foetale hemoglobinespiegels in rode bloedcellen bescherming bieden tegen de complicaties van sikkelcelziekte. Foetaal hemoglobine (HbF) heeft een grotere affiniteit voor zuurstof dan volwassen hemoglobine (HbA) en belemmert polymerisatie.
Het product OTQ923 wordt verkregen door middel van de CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 gentechnologie dat CD34+ hematopoiëtische stam- en progenitorcellen (HSPC’s) aanpast en zich richt op het veranderen van de HBG1- en HBG2-genpromotors (γ-globine) die de expressie van foetaal hemoglobine in de nakomelingen van rode cellen verhoogt.
De onderzoekers voerden een CRISPR-Cas9-onderzoek uit van de promotors van HBG1 en HBG2. Hiervoor verkregen zij CD34+-cellen uit gezonde donoren waarin zij vervolgens met behulp van een elektrisch veld gaten in de celmembranen (elektroporatie) maakten. Hierdoor kon het genetisch sjabloon, het Cas9-eiwit gecombineerd met een van de 72 geleider-RNA’s, in de cel worden ingebracht en ingebouwd.
Vervolgens werd immunokleuring gebruikt om de fractie aan erytroblasten (voorlopercellen) die foetale hemoglobine tot expressie brachten (F-cellen) te bepalen in erytroïde gedifferentieerde nakomelingen. Het gRNA dat resulteerde in het grootste aantal F-cellen (gRNA-68) werd geselecteerd voor klinische ontwikkeling. Daarnaast werden deelnemers met ernstige sikkelcelziekte opgenomen in een multicenter, fase 1-2 klinische studie om de veiligheid en het bijwerkingenprofiel van OTQ923 te beoordelen.
In preklinische experimenten vertoonden CD34+ HSPC’s, afkomstig van zowel gezonde donoren als mensen met sikkelcelziekte, die bewerkt waren met CRISPR-Cas9 en gRNA-68 dat genetische aanpassing op de gewenste locatie had plaatsgevonden en geen off-target mutaties waren waargenomen. De CD34+ HSPC’s produceerden hoge niveaus van foetaal hemoglobine na in vitro differentiatie of xenotransplantatie in immuundeficiëntie muizen. In het onderzoek kregen drie deelnemers autologe OTQ923 na myeloablatieve behandeling en zij werden voor 6 tot 18 maanden gevolgd. Aan het einde van de follow-up periode hadden alle deelnemers engraftment en stabiele inductie van foetaal hemoglobine (foetaal hemoglobine als percentage van totaal hemoglobine, 19,0 tot 26,8%), waarbij foetaal hemoglobine goed verdeeld was in rode bloedcellen (F-cellen als percentage van rode bloedcellen, 69,7 tot 87,8%). Manifestaties van sikkelcelziekte namen af tijdens de follow-up periode.
De gentechnologie CRISPR-Cas9 werd gebruikt voor het aanpassen van de HBG1- en HBG2-gen promotors en bleek een effectieve strategie voor het produceren van foetaal hemoglobine. Behandeling met autoloog OTQ923 bij drie deelnemers met ernstige sikkelcelziekte resulteerde in een langetermijn expressie van foetaal hemoglobine in de rode bloedcellen en tot klinische verbetering van de ernst van de ziekte.
Het onderzoek werd gefinancierd door Novartis Pharmaceuticals; ClinicalTrials.gov nummer, NCT04443907.
Referentie